|
Welcome to the June edition of the GenericsWeb newsletter, now called INNsight to reflect the variety of generics focussed articles and content. This month's drug in focus is Terbinafine, while Anna McKay discusses the implications of the English High Court decision to allow the MHRA authorisation of Alendronate, and Peter Wittner discusses pricing pressures in the generics industry.
If you were forwarded this email and would like to receive future editions of INNsight, become a member by clicking here.
As with all aspects of GenericsWeb - we love to learn from your feedback; please email us your comments about our newsletter format, article content or any other matter.
|
DRUG IN FOCUS: TERBINAFINE |
| 'Drug In Focus' is written by Leighton Howard, a patent information expert with extensive experience in the generic pharmaceutical industry. He has worked within the professional patent information industry and major generic pharmaceutical firms. He is the founder of XIP Pty Ltd, the professional patent search firm responsible for researching all patent data found in GenericsWeb Pipeline Patent Intelligence. Please email any comments or queries. |
|
In August 2005, key patents covering the Antimycotic active ingredient Terbinafine (Lamisil) will expire in European and Australian markets. Based on information contained in the GenericsWeb Pipeline Selector report for Terbinafine, this months Drug In Focus analyses the patent landscape surrounding this product with a view to launching generic equivalents.
The Terbinafine General Information (Table 1) indicates that the innovator brand Lamisil is available in several dosage forms, which can be broadly separated into oral and topical forms. Of these dosage forms, the hydrochloride salt of Terbinafine is used as the active ingredient with the exception of the gel formulation, which contains the base compound.
Table 1: Terbinafine General Information
The Key Patent Indicator (Table 2) shows two patent families claiming the Terbinafine active ingredient. This occurs occasionally where an active ingredient is claimed as one of many possible formulae in a first patent, then a second patent is applied for which claims certain compounds from within these formulae, based on them having superior properties (often referred to as a selection patent). In these cases both the original active ingredient patent and the selection patent must cease to be in-force prior to launch of the corresponding generic equivalent. In the case of Terbinafine, the August 2005 expiry dates in most European territories and Australia is slightly earlier than that of the corresponding US patents, which protect the active ingredient until at least December 2006, this disparity being due to differing patent term legislation and regulatory approval dates between these countries. This US active ingredient patent formed the basis of infringement proceedings by Novartis and a corresponding validity challenge by Dr Reddys Laboratories Ltd, which ultimately resulted in Dr Reddys conceding in September 2004 that the patent is valid and enforceable.
Table 2: Terbinafine Key Patent Indicator
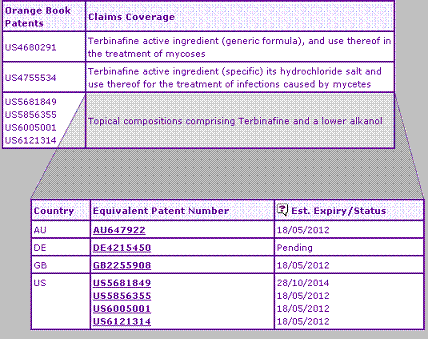
In addition to the molecule patent family, one other constraining patent family is identified by the Key Patent Indicator, relating to topical formulations containing a lower alkanol. Although these formulation patents, which are due to expire from 2012, may represent a barrier to launch of generic topical Terbinafine products, it is not likely that they will prevent generic competition completely.
Such an argument is substantiated by the Patent Risk Analysis, based on comprehensive patent data (details of which are accessible in the corresponding Pipeline Developer subscription). The Patent Category Distribution chart (Figure 1) indicates that patenting activity in relation to this drug is concentrated around formulations. Given that development of a generic oral dosage form is not challenging, it may be assumed that the majority of this activity relates to development of topical formulations. (This is confirmed in the Pipeline Developer report with the identification of over 30 patent families relating to topical formulations of Terbinafine). Also noteworthy is the patenting of different molecular forms of Terbinafine (e.g. salts) to overcome the solubility issues in topical formulations (the hydrochloride salt was disclosed in the active ingredient selection patent).
Figure 1
Furthermore, the Patent Filing Trends chart (Figure 2) shows that the interest in patenting formulations for Terbinafine has been strong since the late 1980s. It is therefore highly likely that a generic formulation of Terbinafine which circumvents the key formulation patents held by the innovator would be possible, given the amount of research in this area. However, caution should be exercised when launching generic topical formulations to ensure that all relevant patents have been considered from an infringement perspective.
Figure 2
In summary, the market for Terbinafine oral tablets has not been well protected by the innovators beyond the life of the active ingredient patents. Later patenting of topical formulations will provide little protection of the topical dosage forms as generic companies are likely to circumvent the innovator patents and perhaps use their own protected formulations to secure a position in the market.
Comprehensive data for patent families relating to Terbinafine, based on professional patent searching, may be accessed by subscribing to GenericsWeb Pipeline Developer reports which include twelve monthly updates to keep you abreast of recently published patents and applications. During the month of June, GenericsWeb are pleased to offer a 20% discount on the standard price of a Pipeline Developer subscription to Terbinafine. GenericsWeb Pipeline Developer and Selector reports are available for any active ingredient upon request.
Click here for more info on GenericsWeb's Pipeline Selector and Pipeline Developer patent intelligence.
Leighton Howard
XIP Pty Ltd
June 2005
l.howard@xip.com.au
Back to top
|
DEVELOPMENTS AT GENERICSWEB |
IGPA Conference, Malta, June 20-22
If you are travelling to the IGPA conference this month, GenericsWeb will have a stand on level 6. Please come for a chat with us or book a time for a demonstration of our Pipeline Patent Intelligence. See the 'events' section in this email for more information on the conference.
If you are new to GenericsWeb you may not know that there is daily generics news archived at our site:
Click here for this month's major generics news headlines
| Contributor Anna McKay specialises in IP Law and Strategy for Pharma companies. As an English solicitor, she won many leading patent cases before the Patents Court, Court of Appeal, EPO, and House of Lords for generic companies. She now works independently, advising companies worldwide on IP exploitation and strategy. |
|
If you would like to contact Anna to give feedback or enquire about her services, please email anna@annamckay.com
Data Protection: High Court allows registration of Alendronate 70mg
High Court judgment
In May the English High Court gave judgment in the action brought by MSD against the MHRAs decision to grant marketing authorisations for alendronate 70mg, a generic version of Fosamax Once Weekly (1). APS (Teva), Generics (UK) and Arrow Generics intervened. The MSD Fosamax case is the latest in a long line of cases before national courts and the European Court of Justice in which the balance of interest between innovators and generic pharmaceutical companies, and the meaning of Directive 2001/83/EC (the Directive) and its precursors have been considered.
The Directive has been amended by Directive 2004/27/EC, which should be implemented by October 2005. Nevertheless, the decision is important for current and past registrations, and as an indication of the way in which the courts think.
Directive 2001/83
The recitals to the Directive provide
experience has shown that it is advisable to stipulate more precisely the cases in which results of pharmacological and toxicological tests or clinical trials do not have to be provided with a view to obtaining authorisation for a proprietary medicinal product which is essentially similar to an authorised product, whilst ensuring that innovative firms are not placed at a disadvantage
there are reasons of public policy for not conducting repetitive tests on humans or animals without overriding cause.
Article 8 of the Directive provides that applications for marketing authorisations have to be accompanied by the following documents:
( c) Qualitative and quantitative particulars of all the constituents of the medical product in usual terminology,
.
(f) Posology, pharmaeceutical form, method and route of administration and expected shelf life
(i) Results of:
- physico-chemical, biological or microbiological tests,
- toxicological and pharmacological tests,
- clinical trials.
Article 10 sets out the essential similarity derogation from this requirement. It provides that an applicant is not required to provide results of toxicological and pharmacological tests or the results of clinical trials if he can demonstrate that the medicinal product is essentially similar to a medicinal product which has been authorized within the Community, in accordance with Community provisions in force, for not less than [six/ten] years and is marketed in a Member State for which the application is made.
However, where the medicinal product is intended for a different therapeutic use from that of the other medicinal products marketed or is to be administered by different routes or in different doses, the results of appropriate toxicological and pharmacological tests and/or of appropriate clinical trials must be provided
It is this derogation which forms the basis of abridged applications for generic pharmaceutical products. The proviso for the derogation to Article 8(3)(i) applies in situations where products are not essentially similar: where the product for which authorisation is sought differs from the product which has been authorised for ten/six years only in particular respects. In certain cases, the applicant may rely upon the original data and submit additional bridging data to cover respects in which there is a difference. This procedure is called the hybrid abridged procedure.
MSD: FOSAMAX: The Background
Fosamax 5mg was authorised in the EU in 1993. Fosamax 10mg in May 1995 and Fosamax Once Weekly in November 2000. MSDs formulation patents on Fosamax were revoked in the UK in January 2003 and in Europea in August 2004. Teva, Generics and Arrow wanted approval for alendronate 70mg, the once weekly preparation. Could the MHRA grant licenses to them for this form under the abridged procedure, even though it had not been marketed in the UK for ten years? MSD claimed that the matter had not been determined by previous cases and that there should be a reference to the ECJ.
Previous Cases
The ECJ has given judgment in three cases relating to the derogation to 8(3)(i). These are the Generics case (2), (new indication), the Novartiscase (3) (Supra-bioavailability no essential similarity) and the APS case (4) (different pharmaceutical form). None of these cases explicitly considered posology (the schedule of dosage: how much and how often a particular dose of medicine should be taken). This, argued MSD, was an issue which had been left open by the ECJ and should be determined by it. The MHRA and intervening generic companies argued that this matter had already been decided, and in order to evaluate whether that was the case, the court considered previous case law.
Generics
The Generics case concerned the grant of a licence for a product for a new indication for which the innovators product had been licensed for less than 10 years. The product had been marketed for more than 10 years for other indications.
The ECJ concluded that:
a medicinal product is essentially similar to an original medicinal product where it satisfies the criteria of having the same quantitative composition in terms of active principles, of having the same pharmaceutical form and of being bio-equivalent, unless it is apparent in the light of scientific knowledge that it differs significantly from the original product as regards safety or efficacy.
Further, A medicinal product that is essentially similar to a product which has been authorised for not less than [6/10] years in the Community and is marketed in the Member State for which the application is made may be authorised, under the abridged procedure
for all therapeutic indications already authorised for that product.
Finally, in relation to new dosage forms and the like the ECJ held that:
A medicinal product that is essentially similar to a product which has been authorised for not less that [6/10] years in the Community and is marketed in the Member State for which the application is made may be authorised, under the abridged procedure
for all dosage forms, doses and dosage schedules already authorised for that product.
The Generics case established that the regulatory authorities could, for an essentially similar product, refer to an innovators data when considering a generic manufacturers application for a marketing authorisation; and, the generic authorisation could cover all of the indications and dosage forms licensed to the innovator as at the date upon which the generic company submitted its application. The case is important not only because the court defined what is meant by essential similarity but also because it shows how the court reconciled the conflicting interest of innovators and generic companies. In particular, it shows the absence of any weight to be given to expenditure and time and costs by innovators in developing their product. It also explains the importance of protection afforded to innovators by Intellectual Property Law to which the data protection given by Article 10 is only an addition.
Novartis
The decision in Generics left open the question of whether an applicant could rely on data supplied by an innovator where the generic product C was not essentially similar to the original product (A) but was essentially similar to a development of that product, product B. Novartis had developed an original product A, Sandimmune, into another product, Neoral, which was not bio-equivalent to Sandimmune.
Generic companies sought authorisation for a product which was essentially similar to Neoral. The court ruled that although bioavailability was not explicitly referred to within the proviso, it was implicit. The generic applicant could rely on the bridging data on which the innovator had relied in relation to Neoral, and the original data in relation to Sandimmune. It was accepted that the products were not essentially similar, and stated that the purpose of the proviso was to allow applicants whose product differed from an existing product in the ways stipulated by the proviso to submit only bridging date.
APS
APS also related to an application for a product which was not in the pharmaceutical form of the original product A, but in the form of the development, product B. Prozac had been authorised in capsule form for more than ten years, and APS sought to market a liquid form of the drug, Fluoxetine, liquid Prozac having been marketed for less than ten years. The court applied the same logic as in Novartis. In other words, it determined that pharmaceutical form, whilst not referred to in the proviso, was implicit within it.
There was an additional argument in APS. It was argued that the generic applicant must apply under the proviso, but the Advocate General and court stated that this was not necessary. The Advocate General concluded that:
There are good reasons against requiring an application in respect of product C to proceed under the proviso. The proviso operates in circumstances where bridging data are required because of a difference between the new product and the earlier product or products to whose data reference is made. Where product C claims essential similarity to product B which is a variant of product A, no addition data are required. There is therefore no need to proceed under the proviso.
MSDs Arguments
MSD sought a reference to the ECJ, on the basis that the question at issue was unresolved. It relied on three distinctions from previous decisions:
- The difference between MSDs original product (Fosamax 5mg and 10mg A) and the development (Fosamax once weekly 70mg B) was one of posology, a difference that had not been the subject of previous decisions.
- Fosamax Once Weekly involved had multiple differences from the reference product A (strength and bioavailability).
- MSD had received authorisation for Fosamax Once Weekly following a full stand alone application, not an application under the hybrid abridged procedure it did not rely on bridging data, and hence the generic applicants should not be entitled to do so.
There was a more fundamental argument, namely that there was no statutory rule for allowing registration through the abridged procedure of products which were not essentially similar. In other words, the whole basis of the ECJs caselaw concerning the proviso was wrong.
The Judge decided that changes in bioavailability were implicit in Novartis. A change in route of administration generally entails different bioavailabilities. Arguably, posology is included in the proviso (different doses). However, the Judge felt that even if the word dose was given a restricted meaning of strength, a change in posology fell implicitly within the proviso: a change in dose generally entails a change in posology. The Judge considered that it would undermine the purpose of Article 10 to provide a further period of protection merely because the innovator had changed the frequency with which doses should be taken by increasing strength. In relation to the second point, it was clear that the fact that Fosamax Once Weekly differed in more than one respect from Fosamax 5mg and 10mg did not take it out of the proviso.
Finally, he stated that it would be wrong if an innovator could acquire a further period of protection merely because the authority had sanctioned a full, as opposed to hybrid abridged, applications. Generic companies could not influence the manner in which innovators applied for product licenses on their developments.
The Judge concluded that generic companies could not be required to provide further data under the proviso where their product was essentially similar to the innovators product B, (the development). Any other construction would not achieve the objective of the recitals of avoiding unnecessary repetitive tests. The only purpose would be to give a further period of protection to innovators but they were already protected by Intellectual Property Rights, and by Article 10.
Finally, the Judge rejected MSDs submissions that the previous ECJ decisions were wrong, in that the proviso only applied where the generic product is essentially similar to the original product. That argument had been made in Novartis, and had failed. Such construction would not achieve the primary purpose of the Directive. Rather, it was inconsistent with the Directive. If the generic product was essentially similar to a development, i.e. was no less safe or efficacious from the development, the only purpose to be achieved by giving the further period of protection was to protect the innovator.
The Judges conclusions as to whether a reference could be made under the proviso to original products and/or developed products are interesting. Applying a purposive approach, it does not matter whether an application is regarded as being under Article 10(1)(a)(iii) or under the proviso. In either case, and the matter to be determined as to whether further tests have to be carried out, and whether or not the product is essentially similar. That is the construction which is required in order to achieve the objectives of the Directive.
To an English lawyer, these comments are interesting. English Law required literal construction, and the intention of legislators was irrelevant. However, European Law (and English Law based on European Law) requires purposive construction. In other words, the purposes of the legislator, the philosophy behind the legislation, are to be taken into account in determining meaning. Hence, it does not really matter whether an application is made under the proviso or not: what is important is that the purpose of the legislator as expressed in the recitals is followed.
Reviewing the case law, the Judge decided that the principles enunciated by the court in Generics, Novartis and APS applied. There was nothing further to be determined by the European Court and a reference was refused. The ECJ had not specifically considered a change in posology before, but the ECJ had considered the principles by which the Treaty was to be interpreted, and the domestic court had to apply these. The MSD case raised no new issues of principle.
No doubt the matter will be appealed. In my view, the decision is correct and a reference should not be allowed. We can expect, however, that tension between the interests of innovators and generic companies will continue.
(1) R (on the application of MERCK SHARP AND DOHME LTD) (Claimant) v THE LICENSING AUTHORITY (ACTING BY THE MEDICINES & HEALTHCARE PRODUCTS REGULATORY AGENCY) (Defendant) & (1) APPROVED PRESCRIPTION SERVICES (UK) LTD (2) GENERICS (UK) LTD (3) ARROW GENERICS LTD (Interested Parties) (2005)
[2005] EWHC 710 (Admin)
(2) Case C-368/96 R v The Licensing Authority established by the Medicines Act 1968 ex parte Generics UK Ltd and others
(3) C-106/01 (on the application of Novartis Pharmaceutical UK Ltd v The Licensing Authority by the Medicines Act 1968
(4) C-36/03 Approved Prescription Services Ltd v The Licensing Authority
Anna McKay
May 2005
www.annamckay.com
| Contributor Peter Wittner has been in the pharmaceutical industry for 30 years. He has worked for the former Evans Medical and then Norton Pharmaceuticals (now part of IVAX) where he was responsible for European Sales & Marketing. After leaving Norton Peter set up his own consultancy in 1993 and operated independently until 1996 when he joined the Indian company Ranbaxy to set up the infrastructure of their new subsidiary. For the last 7 years he has been a consultant in the field of generics. |
|
The Generic Pricing Squeeze
Everyone who works in, or with, the pharmaceutical industry knows that it is often perceived by the public as selling overpriced products of dubious therapeutic value. Actually, that is probably not true since if the leaders of the industry truly appreciated that fact, they might divert some of the industrys mighty marketing and PR prowess towards improving the industrys public image.
So far, however, they have not done so and hence the generally negative perception of the industry remains. The result of this unenthusiastic view is that when governments feel that it is time for them to be seen to be doing something about healthcare costs, beating the pharmaceutical industry over the head about its pricing is seen to be a vote winner.
Subconsciously, many individuals probably feel something along the lines of my poor grandmother is ill and somebody is making a profit out of her illness. And so when the industry is under attack on the issue of prices and profits, few people will rush forward to defend it.
The US pharmaceutical industry in particular leaves itself open to such accusations. Comparisons between prices in the US and elsewhere generally show that it has prices that are significantly higher than in other countries. Some recent evidence to support its critics comes, surprisingly, not from consumer groups or politicians in the US but rather from the UKs Department of Health. In March 2005, it issued its Eighth Report on the PPRS (Pharmaceutical Price Regulation Scheme) that is used to control pharmacy company profits in the UK.
The report includes a table comparing ex-factory prices for a basket of products in the UK to those of other countries. The products involved were the top 150 branded products in the UK that are also generally available elsewhere. Taking the 5-year average for the period 1999 2003 with the UK as 100, the USA came out as 210! Within Europe, Germany was next at 94 followed by Austria at 86; Spain was the lowest at 74. For those who are interested, the full report is available at www.doh.gov.uk/pprs.
At this point, generic observers may say very interesting, but so what? Well, firstly it shows how the US market is ripe for price controls of some sort and secondly when these price controls do arrive they will probably also hit the generic industry as well. If the US Congress for example decides that pharmaceutical process should be marked down by, say, 25% then the starting point for generic introductions after patent expiry will similarly start 25% lower.
What will be worse for the generic industry though is if Congress takes note of what has been happening within certain of the European generic markets that were the subject of last months article. Reference prices at first glance seem to be a relatively good idea for cost control set an upper limit for the price of a particular product and leave it to the pharmacists discretion to decide who should supply the product that he dispenses.
However, the manner in which some countries have sought to implement these schemes have proved quite painful to the generic industry in those markets. When combined with generic substitution as has become widespread across Europe, many governments are effectively putting a price squeeze not just on branded product prices but also on generic prices.
Take the example of Germany where the Aut idem (Latin phrase meaning or similar) regulation is constantly pushing prices lower and lower. It is a hideously complex piece of legislation (memorably entitled Arzneimittelausgaben-Begrenzungsgesetz) that in essence requires a pharmacist to dispense a generic from the bottom third of a reference price group, irrespective of which product in the group has been prescribed. The result of such a scheme is that products priced above this level are excluded until the government publishes the next reference price tables.
The companies whose products are excluded in one round will therefore reduce their prices in order to be included in the lower part of the price table next time around. The inevitable result of this is a constant erosion of prices with the consequent negative impact on the industrys profitability. At the time when the scheme was first introduced in 2002, the local industry body DGV spoke of its fears that this would lead to the demise of some smaller companies with only a handful of survivors which it described in a press release in August 2002 as the last five Mohicans".
It is perhaps understandable that the government of a country that sits in the upper levels of any price comparison would want to introduce a price control mechanism that includes generics. It was however more difficult to understand when something similar happened in Spain whose prices were already low even before the government introduced comparable measures. A combination of reference prices and mandatory substitution is making the newly established Spanish generic industry fearful for its future, and similar control methods are hurting the infant industries in France, Italy and Portugal.
In Spain, Miguel Barbero, the Director General of the Asociación Española de Sustancias y Especialidades farmacéuticas Genéricas (AESEG) spoke of his concerns in a press release of 20th April 2005 following the recent implementation of Annexe III of the Reference Price Order and a 7% generic price reduction. He wrote this situation could lead to the disappearance of the sector by the end of the current legislature".
Perhaps his fears are exaggerated, but perhaps he is correct and the existence of the industry is genuinely under threat.
Whichever is the case, there is a high probability that US legislators are keeping an eye on what is happening in Europe. If they decide to copy some of the cost control systems that have been introduced across Europe the impact on US generic company profits will be painful.
This then leads on to another question. What will happen to members of the US generic industry if they are also subjected to a price squeeze at the same time as ever more low cost producers are entering the US market?
If you have any comments or questions on this article, please contact me on peter@interpharm-consultancy.co.uk
Peter Wittner
May 2005
www.interpharm-consultancy.co.uk
Back to top
|
GENERICS EVENT WATCH JUNE - JULY 2005 |
Cosmo Amman
4-6 June, Amman: Middle East trade fair for cosmetics and pharmaceuticals; technology transfer, agents, distributors and raw materials.
CPhI China
14-16 June, Shanghai: Business meeting place for the leading Chinese and international pharmaceutical ingredient manufacturers and traders. Expecting 11,000+ attendees.
IGPA Pre-Conference Seminars
19 June, Malta: Three seminars, "Legal Framework and Registration Procedures for Generic Medicines in the European Union", Technical licensing requirements for establishing a presence in the North American generic market and IP Strategies for generic drugs in the US market.
IGPA Conference
20-22 June, Malta: Market status: industry and regional perspectives, keys for competing successfully in Generics. Pre-conference seminars on the 19th.
EuroPLX 24
29-30 June, Amsterdam: Global bio/pharma industry partnering event. Proposed orientation of EuroPLX 24 is towards collaborative agreements in clinical and approved, patented drugs.
If you would like us to include details of any additional events that are relevant to the generics industry in our futurre newsletter please email us.
Back to top
. .
Generic Company reports from Espicom Business Intelligence:
Provide an insight into each Generic company, reviewing its current business structure and activities.Recent quarterly and annual financial results for each company are illustrated with comparative figures, charts and a detailed review of the data allowing you to track the company's progress. Information is provided on the company's active product lines and ANDA approvals, along with a review of major developments, such as M&A activity, strategic alliances, and litigation. Companies covered include: Able Laboratories, Andrx, Dr Reddys, Ranbaxy, Sandoz, Stada Arzneimittel. For a full list of all Espicoms Generic Company Reports, including a full list of the companies covered and up-to-date prices of each report, click HERE.
Biogenerics: An Emerging Global Market?
A strategic report from Espicom Business Intelligence published August 2004. There are big rewards for those companies who can overcome the regulatory, legal and technical obstacles related to biogenerics. That is why this critical new report is essential reading for everyone involved or interested in the sector.
|
The GenericsWeb Patent Intelligence Newsletter is available in both HTML-format (text & images) and text-format. If you have any problems with the format, please let us know by emailing: info@genericsweb.com . If this information is not relevant or of interest, please accept our apologies.
Disclaimer: Please note that all information provided in this newsletter is considered accurate at the time of writing but is not guaranteed and may change over time. Opinions expressed in this newsletter are those of the authors and are not intended to represent legal or investment advice. Information should be verified for accuracy and advice should be sought from a suitable professional before acting upon any information contained herein. |
|
GenericsWeb
|
|
GenericsWeb is a division of XIP Pty Ltd responsible for provision of information products dedicated to the generic pharmaceutical industry. If you are interested in contributing to the GenericsWeb newsletter or would like to discuss promotional or partnership opportunities please feel free to contact us.
email: info@genericsweb.com
web: www.genericsweb.com
tel no.: +61 2 9818 6111
fax no.: +61 2 9818 7786
Address: PO Box 202
Balmain NSW 2041 Australia |
|
CONTENT
Patents
DIF: Terbinafine
News
Developments at GWeb
Daily News
Legal Developments
Industry Perspective
Resources
Events Watch
Industry Reports
|